The MD-HMMM-QM workflow for combining MD-based QM and SSNMR data
The MD-HMMM-QM workflow to capture dynamics functional lipid is developed. It enables incorporation of solid state NMR and biochemistry data in collaboration with the Rienstra (UIUC) and Morrissey (Michigan) Labs. We employ a novel combination of the extensive HMMM all-atom MD simulations, comprehensive QM calculations on hundreds of lipid-ion complexes and detailed solid-state NMR spectroscopy on selectively-labeled diluted samples. Agreement between theory and experiment spans multiple spatial scales: 1) MD-based QM-calculated chemical shifts (hence chemical environments) of lipid carbon atoms in two identified long-lived lipid conformations agree with our previous NMR measurements on standard concentration of specifically-labeled samples, 2) characteristic distances and coordination numbers of lipid-ion nanoclusters observed in MD simulations are in exact agreement with NMR measurements on diluted selectively-labeled samples. Importantly and uniquely, use of diluted samples allows us to distinguish inter- and intra-lipid distances that otherwise are not separable (Hallock, …, Pogorelov, 2018).
Highly-Mobile Membrane Mimetic (HMMM) model
The HMMM model is being developed in collaboration with the Tajkhorshid Lab (NIH Center for Macromolecular Modeling and Bioinformatics, University of Illinois at Urbana-Champaign).
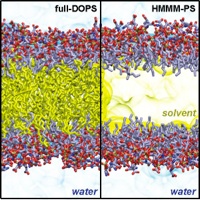
Lateral diffusion of individual lipids in the membrane is a slow process so that a 100 ns molecular dynamics trajectory will lead to few lipid exchanges. We have been developing an all-atom membrane Highly Mobile Membrane Mimetic (HMMM) model (Ohkubo, Pogorelov, et al., 2012) in which conventional lipid tails are shortened and the membrane core is filled with hydrophobic solvent (Fig.). offers the most efficient way to investigate lipid-protein interactions due to model’s enhanced lipid dynamics (1-2 orders of magnitude) without compromising atomistic details. The energetics of amino acid insertion into the interfacial regions of the HMMM model matches that of the full-tail all-atom and coarse-grained lipid representations (Pogorelov et al., 2014). Additionally, the HMMM pressure profiles match well full-tail membrane values from the interface into the core of the membrane, excluding the central 10 A (Qi., et al. 2015). We have also introduced completely hydrophobic in silico designed solvents to replace 1,1-dichloroethane (DCLE), the original solvent with a dipole moment (Vermaas, Pogorelov, Tajkhorshid, 2017). In collaboration with the Im Lab (Lehigh University), the HMMM Builder was implemented in the CHARMM-GUI Portal. Recent applications of the HMMM model to studies of membrane-protein interaction are reviewed in Vermaas et al., 2015 and Baylon et al., 2016.
Hybrid Molecular Dynamics Method
To capture folding the mechanism of point mutants of the fast-folding lambda repressor in an accessible computational time, a hybrid molecular dynamics method was developed and implemented in the NAMD2 software platform using C++ programming language. An all-atom energy potential was augmented by a native-distance biasing Gō-like potential applied only to alpha-carbon atoms Pogorelov & Luthey-Schulten, 2004. The method was successfully used to reveal atomistic details of folding of the fast and slow protein variants. Computational results were compared with experimental data in a companion paper by Yang & Gruebele, 2004.